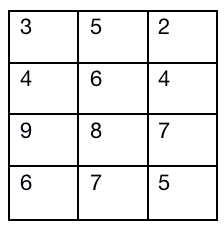
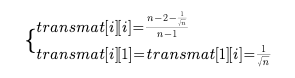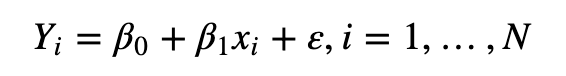quantiles标准化,也就是我将要介绍的最后一个标准化方法,这个标准化方法显然是顾名思义就是根据分位数进行标准化。 该算法在Bioconductor包内的preprocessCore包内,并且是由
阅读更多...
还是和往常一样,首先是要解释一下这个qspline的整体在做一个什么工作。 我们还是要注意这个东西叫做标准化,所以必须是把原数据向某个标准靠拢(映射)。 qspline我没找到网上比较官方的翻译,所以
Bioconductor包之中,有一个标准化方法叫做normalize.AffyBatch.contrasts(),这个方法参照的是一个2001和2003年Magnus Astrand写的两篇文章,其
在affy之中,数据预处理有一个叫做constant normalization的方法。 首先,我要说一个我自己认为的东西。我个人认为这个过程要叫做标准化,而不能叫做归一化。 前一篇文章关于RMA算法
上一篇介绍了RMA算法的背景校正部分。接下来是归一化和最终计算过程。 首先是归一化: 归一化就是将不同组之间的数据统一。因为组之间的数据虽然经过背景校正之后去除了部分噪音。 但是如果某一组自身因为内因
RMA算法是基因芯片数据预处理算法之中一个非常好用而且有效的方法。 本人最近正在学习这个算法,但是发现国内关于这方面的资料是真的很少。 RMA算法分为三个部分, 背景校对 归一化 最终计算表达强度 首
affy包为Bioconductor之中一个用于数据预处理的包. 先解释一些为什么这个包叫做这个名字,这是因为affy其实是一个生产基因芯片的公司, 这个公司做的芯片所产生的数据肯定不是拿过来就能用的
首先,需要简单的申明一下,model.matrix是R语言自身的包,不是bioconductor包里的部分。 所以在理解上,不需要带上生物信息学的知识,也无需生搬硬套bioconductor里的其他函
当删除某一目录下满足条件的文件时,直接使用rm命令即可。 当目录下还有子文件夹时,直接使用rm命令则无法实现删除所有满足要求的文件的功能。 经过上网查阅,可以使用如下命令: 例如我要删除当前目录下所有
rsync -av --exclude /sys --exclude /proc --exclude /database /* /database 排除/sys /proc 以及需要复制的目标位置
作者邮箱二维码